
Approccio diagnostico alla malattia di Gaucher e attuale trattamento
La malattia di Gaucher è una patologia ereditaria autosomica recessiva che, se non diagnosticata o diagnosticata tardivamente, comporta complicazioni devastanti. A causa della natura eterozigote, tale patologia mostra un ampio spettro di presentazioni cliniche; il medico, pertanto, deve essere essere consapevole della possibilità di questa “malattia rara” ma potenzialmente trattabile in pazienti che si presentano con splenomegalia, epatomegalia, anemia, alterazioni scheletriche, ascite o cirrosi da causa non nota (1).
Peraltro, molti pazienti vanno incontro ad una vera e propria “odissea diagnostica” e si stima che il ritardo medio della diagnosi dall’esordio dei sintomi sia di 8–10 anni, quando ormai i danni d’organo possono essere irreversibili (2, 3).
Spesso la diagnosi è sospettata per il rilievo occasionale di cellule di Gaucher in esami bioptici epatici o midollari, richiesti per sospette patologie epatiche o ematologiche (Figura 1).
Figura 1. Tipiche cellule di Gaucher, con citoplasma voluminoso, dal caratteristico aspetto “sgualcito” e nucleo piccolo. |
|---|
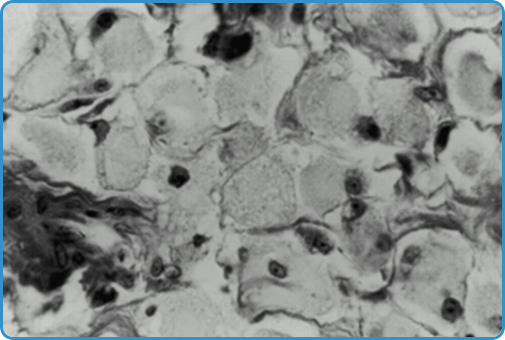 |
Peraltro, pseudocellule di Gaucher possono essere presenti in varie condizioni, come ad es. leucemia linfocitica acuta, mieloma multiplo, linfoma. Il gold standard per la diagnosi pertanto è il test della β-glucosidasi, con la dimostrazione della carenza di attività enzimatica nelle cellule nucleate, ossia leucociti del sangue periferico o fibroblasti cutanei in coltura. L’analisi molecolare genetica è lo standard per la conferma della diagnosi e la ricerca delle mutazioni coinvolte (4).
Il dosaggio di alcuni biomarcatori plasmatici (chitotriosidasi o CCL18) è utile nel follow up, in quanto consente di valutare la risposta alla terapia. (2) Per stabilire la terapia appropriata, tutti i pazienti devono essere sottoposti a valutazione clinica, esami di laboratorio e strumentali che consentono la stadiazione iniziale della malattia (5).
La malattia di Gaucher è la prima malattia da accumulo lisosomiale per la quale è stato sviluppato uno specifico trattamento, la terapia enzimatica sostitutiva (ERT), introdotta nel 1991 (5, 6).
La sostituzione dell’enzima carente, la β-glucocerebrosidasi, con imiglucerasi o con una molecola di più recente introduzione, velaglucerasi alfa, consente la degradazione del substrato glucosilceramide in ceramide e glucosio (2, 5).
Gli obiettivi del trattamento sono la regressione delle alterazioni ematologiche, la riduzione dell’organomegalia e dell’infiltrazione ossea, con la prevenzione delle complicanze conseguenti (Tabella 1) (7).
Tabella 1. Il 25-50% delle pazienti affette da PCOS è sovrappeso/obesa. |
|---|
 |
In uno studio che ha valutato l’efficacia di velaglucerasi alfa nel conseguire precocemente e mantenere gli obiettivi terapeutici nella malattia di Gaucher tipo 1 in 12 pazienti in terapia da almeno 4 anni, tutti i pazienti hanno conseguito i 5 obiettivi terapeutici a lungo termine (anemia, trombocitopenia, epatomegalia, splenomegalia e patologia scheletrica) ed è stato inoltre osservato un continuo miglioramento della densità minerale ossea. I miglioramenti si sono mantenuti anche dopo la riduzione del dosaggio, attuata dopo 15-18 mesi dall’inizio del trattamento (7).
Altri approcci terapeutici in valutazione sono rappresentati dalla terapia di inibizione del substrato (SRT), che consiste nel blocco della formazione di nuove molecole di glucosilceramide, e dalle molecole chaperon, che hanno l’obiettivo di trasportare nei lisosomi l’enzima residuo intrappolato nel reticolo-endotelio (3).
| Bibliografia |
|---|
1. Harmanci O, Bayraktar Y. Gaucher disease: New developments in treatment and etiology. World J Gastroenterol 2008; 14(25): 3968-3973
2. Zimran A. How I treat Gaucher disease. Blood 2011;118(6):1463-1471
3. Cassinerio E, Cappellini GF. La malattia di Gaucher: una malattia rara di interesse internistico. Medicina Italia N. 3/2008
4. Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Pediatric Health, Medicine and Therapeutics 2011; 2: 59–73
5. Martins AM, Ribeiro Valadares E, Porta G et al. , Recommendations on Diagnosis, Treatment, and Monitoring for Gaucher Disease. The Journal of Pediatrics 2009;155 (4, Suppl. 2): S10–8
6. Mucci JM, Rozenfeld P. Pathogenesis of Bone Alterations in Gaucher Disease: The Role of Immune System. Journal of Immunology Research 2015. Article ID 192761
7. Elstein D, Cohn GM, Wang N et al. Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis. 2011;46:119-123
N. 2/2015 - MedTOPICS - Periodico Quindicinale |